













洁净护理单元设计施工
车间内应区分清洁作业区、准清洁作业区和一般作业区。清洁作业区包括湿法工艺的出粉口区域,干法工艺的配料和混合区域、包材暂存间、半成品贮存、充填及内包装车间等。准清洁作业区包括原料预处理车间、其他加工车间和干法工艺的拆包和隧道杀菌区域等。一般作业区包括收乳间、原料仓库、包装材料仓库、外包装车间及成品仓库等。
在线订购产品详情
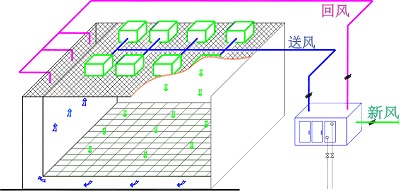
洁净护理单元净化空调系统
洁净护理与隔离单元的防控区和辅助防控区用房,应采用净化空调系统;普通工作区和污物处理区用房可采用送风口无需设过滤器的普通集中空调系统。
洁净护理与隔离单元可采用全新风直流系统,或可全年变新风量运行。
各区宜分开设置空调系统。各类病房空气环境应实行全天候 24 小时全过程控制。
重症监护病房(ICU)及辅房
多床 ICU 空调系统应遵循以下要求:
1. 宜优先采用集中空调系统,送回风口宜采用上送下回布置;
2. 多床 ICU 应在顶棚上设一定数量排风口,风口内应安中效过滤器。
ICU 内的单人组合病房缓冲间对病房和对走廊均必须保持正压或负压。组合病房其他设计参数同于同类重症监护病房。
易感染患者病房及辅房
![]()
![]() 治疗期血液病房的净化空调应采用一对一自循环系统,在病床上方集中送风。
治疗期血液病房的净化空调应采用一对一自循环系统,在病床上方集中送风。
当不具备设置集中净化空调系统或不便在床上方设置集中送风面或有其他特殊原因时,治疗期血液病患者也可采用层流治疗舱(不含冷热盘管),室内应有分散风口送风,达到温湿要求和Ⅰ级用房标准。
治疗期血液病房的净化空调系统应采用独立的双风机并联,互为联锁备用。
易感染患者病房中的单人组合病房缓冲间对病房对走廊均必须保持负压或均保持正压。
重度烧伤病房的净化空调应采用一对一自循环系统,在病床上方集中送风。
恢复期血液病房、普通烧伤病房、过敏性哮喘病房可分别采用该类病房共用净化空调系统,病房应分散布置送风口,上送下回,回风口不应设在门口附近。
负压隔离病房及辅房
负压隔离病房可设非全新风净化空调系统,高危隔离病房应为全新风系统,新风宜集中供应。各病房应独立排风。
采用的非全新风系统应为每间病房独立用本室大部分空气经本室空调机组循环、小部分空气直排的系统。
适宜采用全新风系统的,可全部病房和辅房分别共用一个全新风系统。
由普通工作区进入辅助防控区的缓冲间,对内对外都应为正压。
排风管出口应直接通向室外,伸出屋面或平台的排风立管,其出口宜高出屋面或平台地面 3m 以上。排风机与新风机(或送风机)应联锁,当排风机故障时应立即自动关闭新风机(或送风机)。全新风病房应设备用排风机。
相关规范:
GB 51039-2014 综合医院建筑设计规范
GB 50333-2013医院洁净手术部建筑技术规范
GBT51457-2024《医院洁净护理与隔离单元建筑技术标准》
GB 50591-2010 洁净室施工及验收规范
GB 50736-2012民用建筑供暖通风与空气调节设计规范规范
GB 50243-2016通风与空调工程施工质量验收规范
JGJ312-2013《医疗建筑电气设计规范》
GB55020-2021《建筑给水排水与节水通用规范》
GB50751-2012《医用气体工程技术规范》
相关标签:
100000级清洁作业区; 10万级级清洁作业区
上一篇:中心消毒供应室设计施工
下一篇:洁净手术室设计施工
